Annotating a Genome Using myRAST
It is now possible to get a fairly accurate annotation of a prokaryotic genome in about a day. We honestly believe that the result is often very, very close to what most annotation groups can produce spending months or even man-years. This short tutorial describes our recommended approach to producing a rapid, quite-accurate annotation within about a day (sometimes less for short genomes, and often more for large or diverged genomes).
The approach that we advocate is especially suited to annotating a genome that is quite phylogenetically close to an existing (presumably, well-annotated) genome or set of genomes. In particular, it works well for newly-sequenced pathogen genomes that are close to large groups of already sequenced genomes.
The proposed approach is as follows:
- Run your genome through myRAST (see ***URL***). This produces an initial annotation. There will probably be errors in gene calls, as well as errors in the assigned functions. Those get cleaned up in the next step.
- Once you have produced an initial annotation, you can "walk the genome" looking for genes that need to be deleted, inserted, or just re-annotated.
- Once you have made a quick pass through the genome, we suggest that you export the genome. You will probably wish to do this twice -- once to produce a Genbank formatted version (which can be used by many tools) and a second as a set of tab-separated files suitable for perusing in a tool like Excel.
Running a Genome Through myRAST
Once you start myRAST you should see a screen similar to this
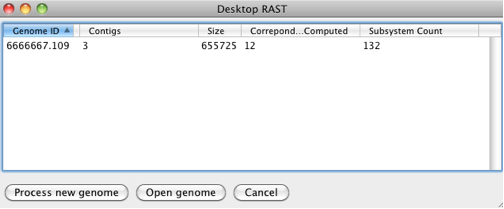
If you click on Process new genome, you will be prompted to pick a file containing the genome to be annotated
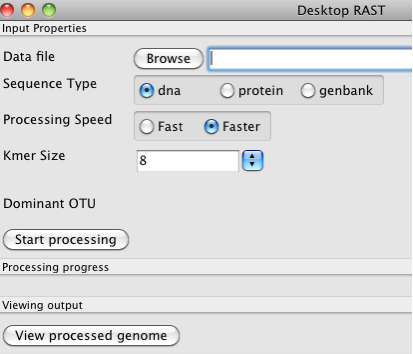
You can take as input a file in Genbank format, a file of contigs in FASTA format, or a file of protein sequences in FASTA format. Normally, you would just specify DNA meaning that you want to annotate some contigs. You need to Browse to get the actual file, and then you click on Start processing to begin building the annotations.
Once you start processing, you will see a "control panel" that looks like
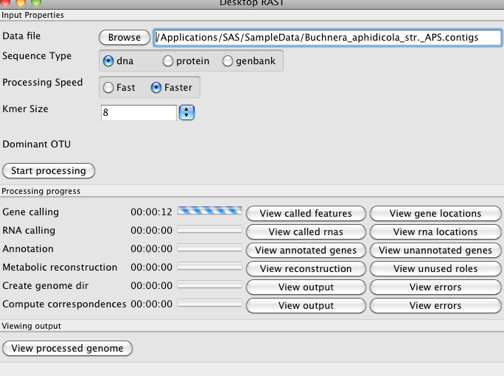
myRAST will go through the annotation steps, you can watch the time it takes, and when it completes you can start perusing the annotations.
Walking your Genome Using myRAST
To begin looking at your annotated genome, you click on View processed genome.
The display shows you what we call a "compare regions display":
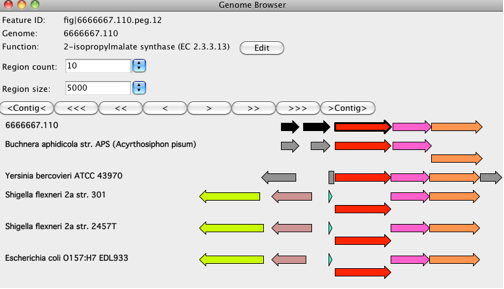
This display shows a region in your newly-sequenced genome (the first line - in this case Buchnera) along with regions from our collection of annotated genomes. The genes (which we call PEGs for "protein-encoding genes") are colored to make it clear which have the same function. All PEGs with the same color have been annotated with the same function. You should think of yourself as focused on one PEG - the one with the bold outline. Hovering over a PEG will give you at least its ID, the contig containing it, begin and end coordinates, and the function assigned to it. PEGs are depicted as arrows. Other features are depicted as rectangles (e.g., in the Yersinia genome, the leucine operon leader is specified as a 133bp "rna" feature). Quite inconsistently, in the Shigella and E.coli genomes, it was annotated as a 28 aa PEG.
You need to spend a little while just figuring out how to interpret a compare regions display, and then try using the navigate buttons:
- ">" and "<" move you 1 gene to the right or left,
- ">>" and "<<" move you a half screen right or left,
- ">>>" and "<<<" move you a full screen right or left, and
- ">Contig>" and "<Contig<" move you to the beginning of the next or previous contig.
What we are proposing you do now, is move through one screen full of compare regions after another, "walking through the genome" to see your annotations and possible errors. This may seem tedious, but for about a day's worth of clicking, you can gain a good sense of the quality and contents of your genome.
Now that you can navigate, let us focus on three important things you can do to change your annotations:
- If you simply wish to change the annotation of a gene, you can "focus" on that gene, click the Edit button, and type in the preferred annotation.
- If you wish to delete the gene, just right-click on the gene and select "Delete feature".
- Finally, you can insert a gene that should have been called, but was not. To do this, position the cursor on the gap where you think the gene belongs, right-click, and select the intergenic region, and then click on a gene from one of the other genomes that you think corresponds to a missing gene that was not called in the intergenic region. This will cause myRAST to try to find an instance of the annotated PEG in the intergenic region. If it finds it, it will mark it in your newly-sequenced genome.
Now we urge you to spend a while moving through your genome to get a feel for what is there and corrections that you can easily make.
Exporting Your Genome from myRAST
-------------------------------
Exporting your annotations from myRAST is fairly straightforward.
There will soon be screenshots here of the procedure.
==========================
Good luck, we hope that you do take the time to try our recommended approach, and we hope that it works as well for you as it does for us.
Suggestions on how we could improve the simple set of tools we provide are welcome.
=======================
